1 INTRODUÇÃO
As proteínas não são moléculas permanentes. Elas estão em processo contínuo de síntese e de degradação, isto é, estão sendo renovadas periodicamente. Cada tipo de molécula de poteína apresenta um tempo de duração denominado de meia-vida da proteína. Entretanto, a concentração de uma determinada proteína é mantida pela síntese desta proteína em quantidade equivalente à de sua degradação.
As proteínas são moléculas formadas de aminoácidos e sua composição é bastante variáveis. Sendo assim, o repertório de aminoácidos originados da degradação das proteínas não é, necessariamente, igual aquele usado para a síntese das moléculas de PTO.
Os aminoácidos excedentes não podem ser armazenados no organismo e, portanto, devem ser oxidados e o nitrogênio de seu grupo amino, excretado.
Os aminoácidos presentes nas células animais originam-se de: aminoácidos derivados da hidrólise das PTO da dieta (proteínas exógenas), aminoácidos derivados da reciclagem das PTO endógenas e aminoácidos essenciais sintetizados a partir de intermediários do metabolismo.
Não armazenamos aminoácidos ou proteínas e, após serem atendidas as necessidades de síntese, os aminoácidos excedentes são consumidos. Isso é feito pela conversão dos aminoácidos em glicose, glicogênio, ácidos graxos, ou então, oxidados a CO2 gerando energia para a célula.
A digestão das proteínas da dieta no intestino e a degradação intracelular de proteínas fornecem o balanço para o organismo.
Função das Proteínas:
- Hormônios e receptores
- Enzimas
- Neurotransmissores
- Proteínas estruturais
- Proteínas contráteis
- Proteínas motoras
- Proteínas transmembranas
- Milhares de peptídeos reguladores, sinalizadores e etc...
1.1 AMINOÁCIDOS
Estrutura básica de um aminoácido
Aminoácidos essenciais: são de alto valor biológico (números acentuados de aminoácidos)
Temos que ingerí-los e são derivados das carnes, peixes e aves, leguminosas.
Aminoácidos não essenciais: não precisamos ingerir. São sintetizados no nosso organismo.
Os aminoácidos são ligados através de
ligações peptídicas, hidratando e liberando H2O.
Ligação peptídica
Ligação peptídica é a ligação do carbono do grupo ácido carboxílico de um aminoácido com o nitrogênio do grupo amina do outro aminoácido.
Cada ligação peptídica libera uma molécula de H2O.
1.2 ESTRUTURAS DAS PROTEÍNAS
Proteína Funcional: tem no mínimo 3 estruturas.
A hemoglobina é formada por 4 cadeias peptídicas e estabilizada pelo íon férrico.
Proteína desnaturada: variação da temperatura, pH mecânico, quando perde a estrutura terciária.
- A proteína ingerida através da dieta é hidrolisada pelo Trato Gastro Intestinal (TGI) em aminoácidos.
- Os aminoácidos ingeridos são utilizados para a síntese de proteína.
- Os aminoácidos restantes são oxidados para fornecimento de energia.
Ovo cru: estrutura tercíária = proteína funcional (estrutura tridimensional ou quaternária)
Ovo cozido: desnaturação da proteína = proteína não funcional
2 DIGESTÃO DAS PROTEÍNAS DA DIETA
A digestão das proteínas começa no
estômago, o pH é de 2 a 3 (ácido), o estômago secreta o
suco gástrico (que é liberado na presença de proteína) e que contém o
ácido clorídrico (HCl) e a
proenzima pepsinogênio. O
ácido clorídrico desnatura a proteína que não está desnaturada e ativa o
pepsinogênio (na presença de proteína) formando a
pepsina. A ativação da pepsina ocorre através do rompimento de alguns aminoácidos do pepsinogênio. Sobre então a pepsina ativa pois se encontra sem os aminoácidos indesejáveis.
A função da pepsina é degradar as ligações peptídicas.
Pepsinogênio (liberado pelo suco gástrico) --- HCl ---- pepsina
Pepsinogênio ---- pepsina ---- pepsina
Ou seja, o pepsinogênio também é ativado por outras moléculas de pepsina já ativadas.
A
pepsina vai degradar a proteína no estômago liberando
polipeptídeos. Então, esses polipeptídeos chegam no
intestino delgado e como consequência é liberado o
suco pancreático. Ao ser liberado, o
suco pancreático (bicabornato + zimogênios) deixa de ser ácido (aumenta o pH), junto com os zimogêneos (
tripsinogênio e
quimiotripsinogênio).
O
tripsinogênio perde alguns aminoácidos formando a
tripsina e o
quimiotripsinogênio virando a
quimotripsina, degradando os polipeptídeos, liberando os aminoácidos.
Tripsinogênios e quimiotripsinogênios são zimogêneos inativos.
Tripsina e quimiotripsina são zimogêneos ativos.
Os aminoácidos quebrados vão para o fígado para a síntese protéica e os em excesso que não forem utilizados vão ser degradados no fígado também.
Resumo:
A proteína é ingerida através da boca
Chega no estômago e então o HCl + pepsinogênio vai desnaturá-la
O HCL ativa o pepsinogênio liberando a pepsina
Novos pepsinogênios são ativados pelas pepsinas
Proteínas + pepsinas = polipeptídeos
No intestino delgado, há liberação do suco gástrico (bicabornato + zimogênios inativos)
Tripsinogênios e quimiotripsinogênios vão catalisar a degradação das ligações polipeptídicas liberando aminoácidos.
3 DEGRADAÇÃO DE PROTEÍNAS ENDÓGENAS OU
DEGRADAÇÃO INTRACELULAR DE PROTEÍNAS
Inúmeros processos fisiológicos são controlados pela variação da concentração de proteínas específicas. Isso é devido à reciclagem intracelular das proteínas. Algumas proteínas estão presentes somente em uma determinada fase do ciclo celular; outras, como as enzimas reguladoras das vias metabólicas, precisam estar presentes e suas concentrações ajustadas às variações das condições isto é, com alterações em sua composição ou função, devem ser degradadas para que não ocorra comprometimento à homeostasia celular e, assim, o balança protéico intracelular é mantido.
As proteínas intracelulares devem ser marcadas para degradação pela ligação covalente com a
ubiquitina. A
ubiquitina liga-se às proteínas destinadas a degradação.
O complexo protéico denominado
proteossoma reconhece as proteínas ligadas a
ubiquina e as hidrolisam, liberando a ubiquina livre.
As proteínas reconhecidas pelo
proteossoma são degradadas até
aminoácidos e
peptídeos.
A degradação ocorre em dois processos:
1 - O primeiro é realizado por proteases lisossômicas, denominadas
catepsinas. Degrada principalmente proteínas de membranas, proteínas extracelulares e proteínas de meia-vida longa.
2 - O segundo processo ocorre no citosol e acontece com a mediação de uma proteína chamada
ubiquitina. O primeiro passo da degradação é a ligação da ubiquina à proteína em uma sequência de reações que ocorrem com gasto de ATP. O destino da proteína ligada à ubiquitina é a degradação. A proteína ubiquitinada interage com um grande complexo proleolítico, o
proteassoma, que hidrolisa todas as ligações, liberando os aminoácidos.
As proteínas reconhecidas pelo proteossoma são degradadas a aminoácidos e peptídeos.
A proteína será degradada se foi produzida errada ou se o prazo de validade dela acabou. Neste caso ocorre a degradação endógena de proteínas.
Então,
Os aminoácidos são absorvidos e serão utilizados para a síntese protéica nos tecidos. Os excessos serão degradados. O excesso de aminoácidos será sempre degradado.
A síntese de proteínas ocorre no RER (Retículo Endoplasmático Rugoso), onde ocorra a transformação da proteína. No Complexo de Golgi será analisada - verifica se a proteína está certa - se não estiver, será degradada = sistema de reparo.
Ou seja, o complexo de golgi, percebe que a proteína está errada, ela vai ser ubiquitinada e será degradada. Os aminoácidos poderão ser utilizados para outra síntese ou serão degradados. A ubiquitina libera a proteína para ser degradada. E o proteassoma vai degradá-la.
Quando o Complexo de Golgi analis a proteína, ela poderá seguir três caminhos:
- Se a síntese está correta: ela será utilizada pelo organismo
- Se a síntese não está correta: será degradada via lisossomo
- Se a vida útil dela está vencida: será marcada pelas ubiquitinas (ficando poliubiquitinadas) e então será degradada por proteassoma.
Os aminoácidos se originam de: dieta, síntese endógena e degradação de proteína.
Os aminoácidos vão servir para: síntese de proteína, síntese de outras moléculas, degradação (excessos).
Destino dos aminoácidos: síntese de proteína ou oxidação para formação de ATP (fígado/músculo)
4 OXIDAÇÃO DOS AMINOÁCIDOS
O grupo amino garante que o aminoácido seja degradado (oxidado). Quando é removido o grupo amino, o nitrogênio pode ser incorporado em outros compostos ou excretado e os esqueletos carbônicos convertidos em glicose, glicogênio, ácidos graxos ou oxidados a CO2.
Ou seja, sofrendo uma transaminação (retirada do grupo amino para o alfa-cetoglutarato) possiblitará que os aminoácidos sejam utilizados como fonte de energia.
4.1 TRANSAMINAÇÃO
Ocorre em todas as células
Remoção do grupo amino dos aminoácidos
No fígado, o primeiro passo no catabolismo dos aminoácidos é a remoção de seus grupos amino por transaminações são o principal processo para remover o nitrogênio dos aminoácidos.
O
grupo amino é transferido do aminoácido original para o
alfa-cetoglutarato, formando
glutamato, enquanto o aminoácido original é convertido em
cetoácido correspondente. Essa reação é catalisada por um grupo de enzimas denominada
transaminases.
O
cetoácido alfa-cetoglutarato desempenha um papel central no metabolismo dos aminoácidos, recebendo os grupos amino de outros aminoácidos e transformando-se em
glutamato. O grupo amino do
glutamato formado pode ser transferido, por reação de transaminação, ao
oxaloacetato, produzindo
aspartato e regenerando o
alfa-cetoglutarato.
Todos os vinte aminoácidos protéicos, exceto a lisina e a trionina, transaminam com o alfa-cetoglutarato, dando os produtos glutamato e o cetoácido correspondente ao aminoácido transaminado. Para a maioria das reações de transaminação, o alfa-cetoglutarato e o glutamato servem como um dos pares alfa-ácido-aminoácido.
O grupo amino deve ser eliminado porque é é tóxico.
As aminotransferases da maioria dos tecidos de mamíferos utilizam o alfa-cetoglutarato como aceptor do grupo amino, formando glutamato; podem reagir também, embora com afinidade menor, com o oxaloacetato, que é convertido em aspartato.
Ou seja, o glutamato é o primeiro reservatório temporário do grupo amino e o aspartato é o segundo depositório do grupo amino.
Depois que a transaminase glutâmica pirúvica (TGP) retira o grupo amino e passa para o glutamato, o glutamato pode seguir outro caminho de transaminação que ocorre na conversão em aspartato, quem faz essa reação é a transaminase glutâmica oxalacética (TGO) que retira o grupo amino do glutamato e transfere para o oxaloacetato formando aspartato.
TGP: dá origem a piruvato e glutamato
O aminoácido reage com alfa-cetoglutarato formando alfa-cetoácido + glutamato
TGO: dá origem a alfa-cetoglutarato e aspartato
O glutamato reage com oxaloacetato formando alfa-cetoglutarato + aspartato
As aminotransferases são as responsáveis em catalisarem reações de transaminação
Importante: o glutamato é o aminoácido mais abundante na corrente sanguínea pois todos os aminoácidos degradados irão liberar glutamato pela transaminação.
4.2 DESAMINAÇÃO
Seguindo a outra condição do glutamato - desaminação. Vai então, ser desaminado oxidativamente pelo glutamato desidrogenase liberando o grupo amino como amônia livre e o alfa-cetoglutarato. Nesta reação, as coenzimas NAD+ ou NADP+ podem servir como co-fatores. A desaminação oxidativa do glutamato ocorre na mitocôndria da maioria das células e como reação reversível, pode incorporar amônia ao glutamato ou liberar amônia a partir de glutamato.
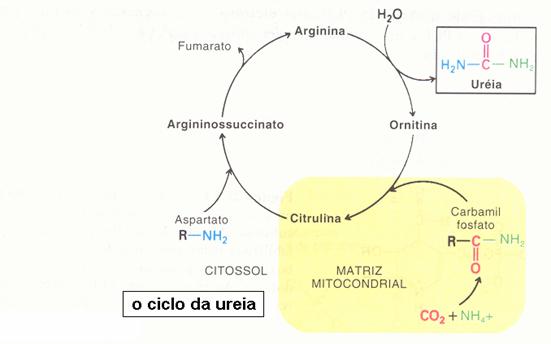
As reações de desaminação oxidativa ocorrem, principalmente, no fígado e no rim. Elas fornecem alfa-cetoglutarato que podem entrar nas vias centrais do metabolismo energético e a amônia ser utilizada como substrato para a síntese de ureia.
Perde o grupo amino na forma de amônia e restaura o alfa-cetoglutarato. Ou seja, o glutamato vai para a corrente sanguínea, chega no fígado, e então, ocorre a desaminação dentro do fígado. As enzimas que catalisam essa reação estão presentes apenas no fígado. Não ocorre a desaminação antes da transaminação porque tem que estar em glutamato para ser desaminado.
Depois que a amônia é liberada, ela vai para o ciclo da uréia.
Se aumenta os aminoácidos, aumenta também alfa-cetoglutarato. Se não tiver a mesma quantidade de alfa-cetoglutarato o grupamento amino vai ser transferido para o glutamato, ficando então com dois grupos amina (glutamina). No fígado, a glutamina vai perder o grupo amino formando o glutamato.
Glutamina = NH3 + glutamato